Introduction
genal is a Python toolkit for common GWAS-derived workflows:
Preprocess GWAS summary statistics into a consistent SNP table (column validation, allele checks, optional filling of missing
SNP/CHR/POS/EA/NEA/SE/Pusing reference data, and computation of per-variant F-statisticFSTAT).Select instruments via LD clumping (PLINK 2).
Compute PRS on individual-level genotype data (PLINK 2), with optional proxy SNP support.
Run two-sample MR (multiple estimators + sensitivity analyses), with plotting helpers.
MR-PRESSO (parallel implementation) for outlier detection and distortion testing.
Colocalization using approximate Bayes factors (ABF) to assess whether two signals likely share a causal variant.
Utilities: liftover between builds, GWAS Catalog annotation, gene-window filtering, allele-frequency updates from a reference panel.
genal is centered around a single class, genal.Geno, which wraps a pandas.DataFrame of SNP-level data and provides end-to-end workflows.
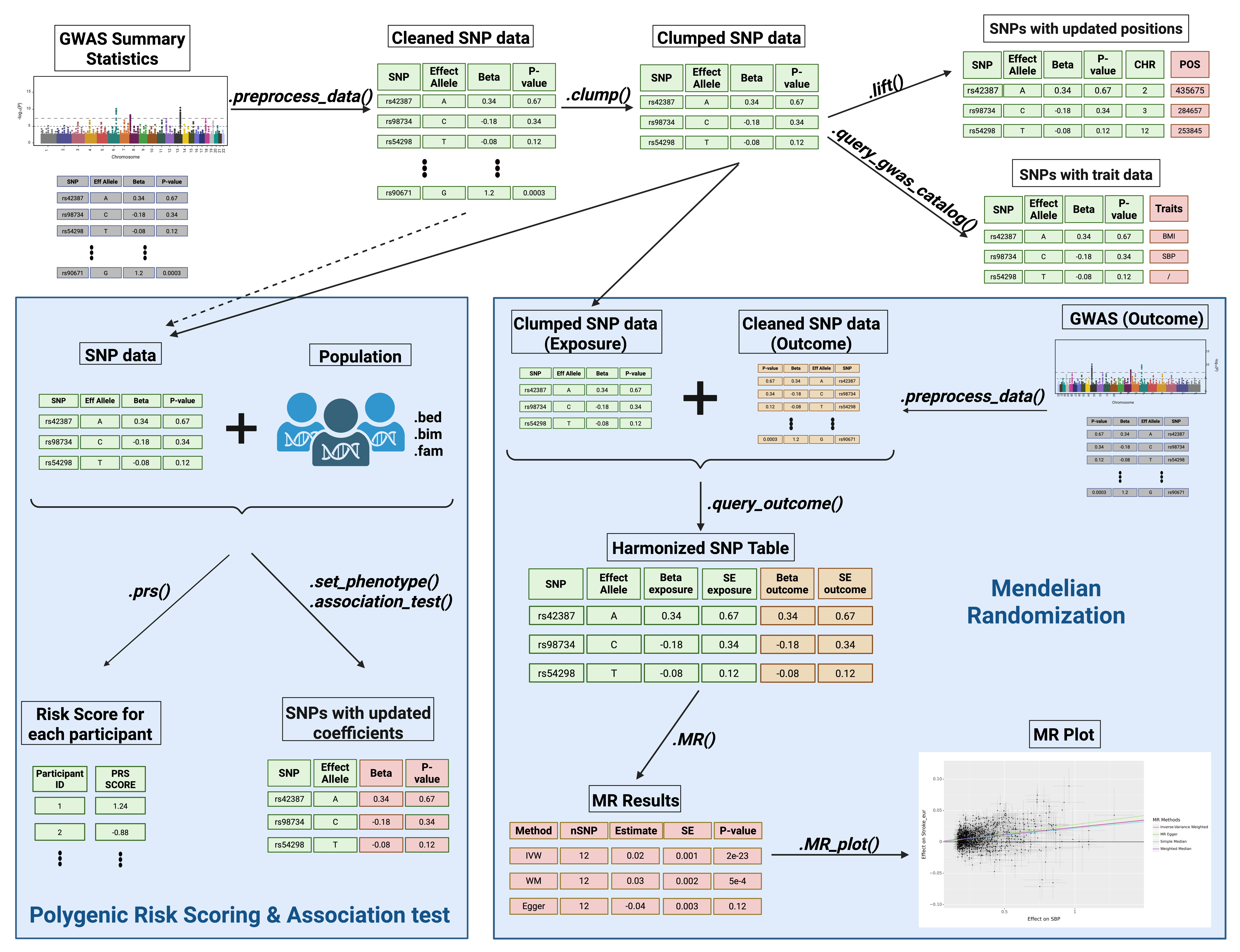
High-level pipeline (GWAS → instruments → PRS/MR).
Design notes
Geno.datais always the “source table”. Most methods either modify it in place or return a newGeno(see Core concepts).Operations that require LD or genotypes delegate to PLINK 2 (clumping, proxy search, scoring, association testing, allele-frequency updates).
Reference data is cached under
~/.genal/by default (config + downloaded panels). See Setup.
When to use genal (and when not)
Use genal when you already have:
GWAS summary statistics (exposure and/or outcome), and/or
PLINK genotype files for a target cohort.
genal does not try to replace a full genotype QC pipeline; it assumes you provide reasonable inputs (or you call preprocess_data() to enforce basic validity).